
Portare un dispositivo medico sul mercato è uno dei percorsi di sviluppo prodotto più complessi e impegnativi in qualsiasi settore. Ogni decisione, dal primo schizzo di un concetto agli anni di monitoraggio post-commercializzazione, ha un impatto diretto sulla sicurezza del paziente. È proprio questa responsabilità a rendere il processo così rigorosamente strutturato.
Il processo di sviluppo dei dispositivi medici è una sequenza articolata in nove fasi, con passaggi strutturati (“stage-gated”), che accompagna un dispositivo dal concetto clinico al mercato commerciale. Comprende la fattibilità, la progettazione, la verifica, i test preclinici, la sperimentazione clinica, la sottomissione regolatoria, la validazione della produzione, il lancio commerciale e la sorveglianza post-commercializzazione lungo l’intero ciclo di vita. Nessuna fase è opzionale: ciascuna genera evidenze su cui si basa la fase successiva.
Il processo è regolato da un insieme di standard e normative internazionali: ISO 13485:2016 per il sistema di gestione della qualità, ISO 14971:2019 per la gestione del rischio, IEC 62304 per il software, IEC 62366-1 per l’ingegneria dell’usabilità e ISO 10993 per la biocompatibilità. Negli Stati Uniti, l’aggiornamento della Quality Management System Regulation (QMSR) della FDA, in vigore dal 2 febbraio 2026, integra direttamente la ISO 13485:2016 nel 21 CFR Part 820. Nell’Unione Europea, il Regolamento sui dispositivi medici (MDR 2017/745) definisce il quadro di valutazione della conformità per la marcatura CE.

La classificazione del rischio del dispositivo determina il livello di evidenza richiesto in ogni fase. Un dispositivo impiantabile di Classe III richiede anni di dati clinici prospettici, una sottomissione PMA completa e obblighi di follow-up clinico post-commercializzazione (PMCF) per tutta la vita del dispositivo. Un dispositivo di Classe II, a rischio inferiore, segue un percorso più breve, ma non per questo meno rigoroso.
La maggior parte dei dispositivi richiede tra i 3 e i 7 anni per passare dal concetto all’autorizzazione alla commercializzazione. I team che comprendono l’intero processo fin dall’inizio lo integrano nel proprio piano di sviluppo sin dal primo giorno, costruendo tempistiche, budget e strategie regolatorie in base alle evidenze effettivamente richieste.
Questa guida analizza tutte e nove le fasi, aggiornate al più recente contesto normativo per il periodo 2025–2026.
Qual è il processo di sviluppo dei dispositivi medici?
Il processo di sviluppo dei dispositivi medici è la sequenza strutturata di fasi che un fabbricante segue per portare un dispositivo dal concetto iniziale al mercato commerciale e per mantenerne la sicurezza durante l’intero ciclo di vita operativo.
Ogni fase si basa sulla precedente. La progettazione alimenta la verifica. La verifica alimenta le evidenze cliniche. Le evidenze cliniche supportano la sottomissione regolatoria. Se una fase viene saltata o accelerata eccessivamente, i problemi si amplificano durante i test, nella revisione regolatoria e, soprattutto, negli esiti per i pazienti.
Il processo è regolato da standard internazionali e quadri normativi, tra cui ISO 13485:2016, ISO 14971:2019, IEC 62304, il 21 CFR Part 820 della FDA (aggiornato nell’ambito del QMSR, in vigore da febbraio 2026) e il Regolamento (UE) 2017/745 sui dispositivi medici (MDR).
La complessità del dispositivo e la sua classificazione di rischio determinano la durata del percorso. La maggior parte dei dispositivi medici richiede tra i 3 e i 7 anni per passare dal concetto all’autorizzazione alla commercializzazione.
L’intero processo di sviluppo comprende nove fasi:
- Concetto e fattibilità — definizione del problema clinico, valutazione della fattibilità tecnica, impostazione del sistema di gestione della qualità e identificazione del percorso regolatorio.
- Progettazione e sviluppo — trasformazione delle esigenze dell’utente in specifiche ingegneristiche verificate, gestione iterativa del rischio e documentazione di ogni decisione progettuale.
- Verifica e validazione — conferma che il dispositivo sia realizzato correttamente (verifica) e che soddisfi le esigenze cliniche reali (validazione).
- Test preclinici — valutazione della sicurezza biologica, della sterilizzazione, della sicurezza elettrica e della durata di conservazione prima di qualsiasi esposizione umana.
- Sperimentazione clinica — generazione di dati su sicurezza e prestazioni nell’uomo in condizioni controllate, sotto supervisione normativa (IDE e comitati etici/IRB).
- Sottomissione e approvazione regolatoria — raccolta di tutte le evidenze in un dossier formale per l’autorizzazione FDA, la marcatura CE o altre approvazioni equivalenti.
- Industrializzazione e validazione dei processi produttivi — dimostrazione che i processi produttivi garantiscono in modo costante dispositivi conformi alle specifiche anche su scala commerciale.
- Lancio commerciale — coordinamento delle registrazioni, della distribuzione, della formazione clinica e dei meccanismi di rimborso prima che il dispositivo raggiunga il paziente.
- Sorveglianza post-commercializzazione — monitoraggio continuo della sicurezza e delle prestazioni nel mondo reale per tutta la vita commerciale del dispositivo.
Ogni fase prevede input, output, obblighi regolatori e punti di controllo della qualità ben definiti. Nessuna fase è opzionale. Le autorità regolatorie negli Stati Uniti, nell’Unione Europea e nella maggior parte dei mercati globali richiedono evidenze documentate che dimostrino che ogni fase sia stata completata con rigore, non semplicemente dichiarata.
Il rigore di questo processo esiste per una sola ragione: i pazienti che dipendono da questi dispositivi non hanno alcun margine per errori o scorciatoie.
Quali sono le fasi dello sviluppo dei dispositivi medici?
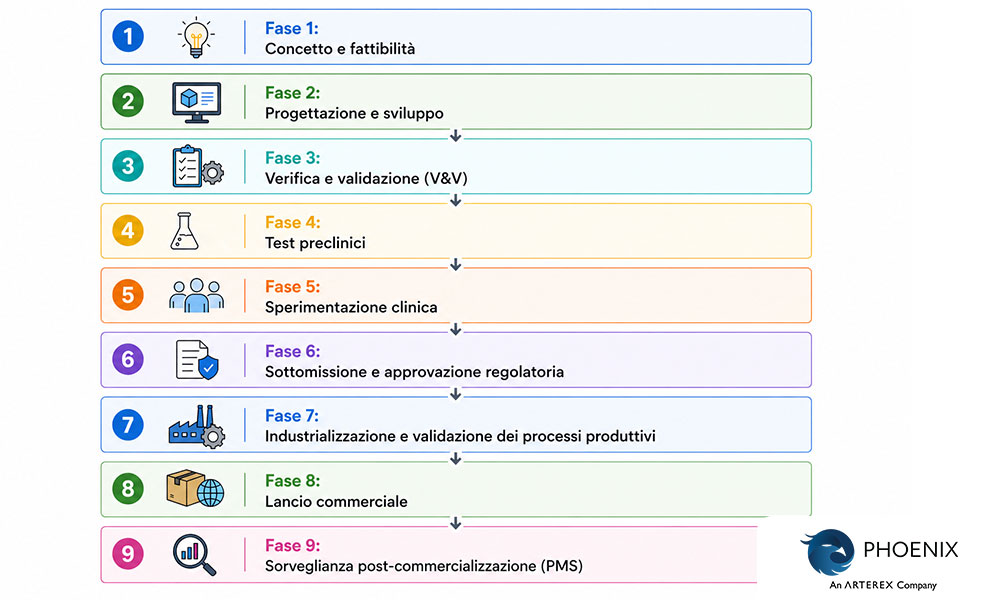
Lo sviluppo dei dispositivi medici segue un processo strutturato, articolato in fasi sequenziali (“stage-gated”), progettato per garantire sicurezza, efficacia e conformità normativa prima che un dispositivo raggiunga i pazienti. Ogni fase si basa sulla precedente, partendo da un concetto iniziale fino ad arrivare a un prodotto pronto per il mercato, soggetto a una sorveglianza post-commercializzazione continua.
I riferimenti normativi sono stati aggiornati per riflettere il Quality Management System Regulation (QMSR) della FDA (21 CFR Part 820), in vigore dal 2 febbraio 2026, e il Regolamento (UE) 2017/745 sui dispositivi medici (MDR).
Fase 1: Concetto e fattibilità
Ogni dispositivo nasce da un problema. Un clinico individua una lacuna nel flusso di lavoro. Un ingegnere riconosce un limite nelle tecnologie esistenti. Un gruppo di advocacy dei pazienti evidenzia un bisogno non soddisfatto. Questa osservazione diventa il punto di partenza di un nuovo concetto di dispositivo.
In questa fase, team interdisciplinari — che includono ingegneria, ambito clinico, affari regolatori e funzioni commerciali — collaborano per valutare se l’idea sia tecnicamente realizzabile e commercialmente sostenibile. L’obiettivo non è ancora costruire il dispositivo, ma determinare se valga la pena svilupparlo.
La norma internazionale ISO 14971:2019 definisce terminologia, principi e un processo completo per la gestione del rischio dei dispositivi medici. Essa stabilisce un quadro sistematico per identificare i pericoli associati ai dispositivi, stimare e valutare i rischi correlati, controllarli e monitorare l’efficacia delle misure di controllo lungo l’intero ciclo di vita del dispositivo. La gestione del rischio inizia qui, nella fase di concetto, non durante i test.
Attività principali:
- Analisi dei bisogni — identificazione del problema clinico, della popolazione target e del bisogno non soddisfatto con precisione. Definizioni vaghe portano a soluzioni progettuali inefficaci.
- Pianificazione del percorso regolatorio — comprendere quale percorso normativo si applica al dispositivo è la prima decisione critica che influenza tempistiche di sviluppo, costi e strategia di accesso al mercato. I team che identificano precocemente la classificazione FDA e la classe di rischio nell’UE sviluppano piani più efficienti.
- Valutazione iniziale del rischio — individuazione dei potenziali problemi di sicurezza secondo ISO 14971:2019, inclusi i pericoli legati all’uso previsto, all’uso scorretto prevedibile e alla popolazione di pazienti.
- Valutazione tecnologica — analisi delle soluzioni esistenti, dei brevetti, dello stato dell’arte e della letteratura scientifica per confrontare il concetto proposto con le tecnologie disponibili.
- Prototipazione di fattibilità — realizzazione di modelli preliminari (proof of concept) per testare i meccanismi fondamentali. Non si tratta di prototipi regolatori, ma di strumenti di apprendimento.
- Definizione del sistema di gestione della qualità (QMS) — la norma ISO 13485:2016 rappresenta lo standard internazionale per i sistemi di gestione della qualità dei dispositivi medici, adottato globalmente dalla maggior parte dei fabbricanti. Il QMS definisce procedure, controlli, modelli e SOP che regolano tutte le attività successive di sviluppo.
- Sviluppo del business case — valutazione della dimensione del mercato, del sistema di rimborso e del posizionamento competitivo. Anche un dispositivo tecnicamente eccellente, senza un percorso di rimborso sostenibile, incontra notevoli difficoltà commerciali.
Uno studio di fattibilità strutturato consente di stabilire se un concetto meriti ulteriori investimenti. I team che saltano questa fase spesso scoprono problemi fondamentali di progettazione durante le fasi di verifica e validazione, ovvero nel momento più costoso per individuarli.
Fase 2: Progettazione e sviluppo
Questa fase è il momento in cui le idee diventano ingegneria. I team trasformano gli output della fase di fattibilità in un progetto di dispositivo definito e documentato. Gli input di progettazione — tra cui esigenze dell’utente, uso previsto e requisiti normativi — vengono convertiti in modo sistematico in output di progettazione, come specifiche tecniche, disegni ingegneristici, selezione dei materiali e architettura software.
Il Quality Management System Regulation (QMSR), entrato in vigore il 2 febbraio 2026, modifica i requisiti delle current good manufacturing practices (CGMP) previsti dal 21 CFR Part 820, incorporando per riferimento lo standard internazionale ISO 13485:2016. In base al QMSR, le attività di progettazione negli Stati Uniti sono ora disciplinate dalla clausola 7.3 della ISO 13485:2016, allineandosi allo standard globale già adottato dalla maggior parte dei mercati.
Ciò significa che le aziende che operano a livello globale non devono più mantenere sistemi di qualità separati per il mercato statunitense e per quelli internazionali. Questa convergenza riduce le duplicazioni e rafforza l’approccio alla progettazione basato sul rischio tra le diverse giurisdizioni.
Con il QMSR, la FDA adotta terminologia e struttura della ISO, inclusa l’espressione “progettazione e sviluppo”, precedentemente nota come “controlli di progettazione”. L’obiettivo regolatorio rimane invariato: i dispositivi devono essere progettati in modo controllato per garantire risultati sicuri ed efficaci in modo coerente.
Un cambiamento rilevante nel nuovo contesto QMSR riguarda il maggiore rigore con cui la FDA applica i requisiti di gestione del rischio previsti dalla ISO 13485, fortemente basati sulla ISO 14971 come parte integrante della progettazione e dello sviluppo. La ISO 13485 richiede che gli output delle attività di gestione del rischio siano utilizzati come input nel processo di progettazione e sviluppo.
Attività principali:
- Documentazione di input e output di progettazione — trasformazione delle esigenze dell’utente in requisiti tecnici specifici, misurabili e verificabili. Nei progetti complessi, questa fase può rappresentare fino al 30% del tempo totale: gli input devono essere completi, non ambigui e verificati tramite protocolli di verifica.
- Sviluppo di prototipi — realizzazione di prototipi funzionali per testare le prestazioni e individuare iterativamente eventuali lacune progettuali.
- Gestione del rischio — applicazione della ISO 14971:2019 per identificare i pericoli, valutare i rischi, implementare misure di controllo e documentare l’accettabilità del rischio residuo a ogni iterazione progettuale.
- Sviluppo software — applicazione della norma IEC 62304 per il ciclo di vita del software dei dispositivi medici, inclusa la classificazione del software, la progettazione dell’architettura, l’implementazione delle unità e la gestione delle modifiche.
- Pianificazione della biocompatibilità — selezione dei materiali conformi ai requisiti della ISO 10993, in funzione del tipo di contatto con il corpo, della durata e della popolazione di pazienti.
- Fascicolo di progettazione e sviluppo (Design and Development File, DDF) — precedentemente noto come Design History File (DHF) nel vecchio QSR, ora denominato DDF nel contesto QMSR. Questo documento dinamico registra tutte le decisioni progettuali, le revisioni e le evidenze lungo l’intero ciclo di sviluppo.
- Revisioni di progettazione — punti di controllo strutturati e documentati che coinvolgono stakeholder di ingegneria, clinici, qualità e affari regolatori per valutare la maturità del progetto e risolvere le criticità prima di avanzare alla fase successiva.
La fase di progettazione stabilisce le basi tecniche per tutto ciò che segue. Input di progettazione incompleti, una gestione del rischio insufficiente o decisioni non documentate in questa fase generano problemi a cascata nelle fasi di verifica, validazione e revisione regolatoria.
Fase 3: Verifica e validazione (V&V)
La verifica e la validazione sono due processi distinti ma complementari che confermano che un dispositivo sia stato progettato correttamente e che risponda alle esigenze reali.
La verifica risponde alla domanda: abbiamo realizzato correttamente il dispositivo? Essa conferma che gli output di progettazione soddisfano gli input di progettazione attraverso test oggettivi e riproducibili, tra cui prove di laboratorio (bench testing), ispezioni dimensionali e valutazioni delle prestazioni rispetto alle specifiche tecniche.
La validazione risponde alla domanda: abbiamo realizzato il dispositivo giusto? Essa conferma che il dispositivo finale soddisfi le esigenze degli utenti e l’uso previsto in condizioni cliniche reali o simulate, inclusa l’interazione degli utenti con il dispositivo.
Nell’Unione Europea, il MDR richiede ai fabbricanti di dimostrare la conformità ai Requisiti Generali di Sicurezza e Prestazione (GSPR), che includono intrinsecamente attività robuste di verifica e validazione. La documentazione tecnica deve dimostrare che gli output di progettazione soddisfano le specifiche e che il dispositivo è clinicamente sicuro ed efficace.
Attività principali:
- Sviluppo e validazione dei metodi di prova — garantire che le procedure di test siano affidabili, riproducibili e adeguate allo scopo prima di generare evidenze regolatorie.
- Prove di laboratorio (bench testing) — valutazione delle prestazioni meccaniche, elettriche e funzionali in condizioni controllate rispetto a criteri di accettazione definiti.
- Test in condizioni simulate d’uso — valutazione del dispositivo in ambienti che replicano l’uso clinico reale, includendo popolazioni di utenti e scenari d’uso realistici.
- Fattori umani / ingegneria dell’usabilità — le autorità regolatorie considerano oggi i fattori umani un elemento essenziale per la sicurezza del paziente. La FDA richiede dati di validazione dei fattori umani nelle sottomissioni pre-market per molti dispositivi. Gli organismi notificati nell’UE richiedono un Usability Engineering File (UEF) secondo la norma IEC 62366-1 nell’ambito della valutazione di conformità MDR. Errori come un’infermiera che programma in modo errato una pompa di infusione o un paziente che interpreta male un display sono modalità di guasto reali che l’ingegneria dell’usabilità mira a prevenire.
- Valutazioni formative e sommative — le valutazioni formative sono iterative, condotte durante lo sviluppo e utilizzate per migliorare il design. Le valutazioni sommative sono effettuate sul prodotto finale per dimostrare che il dispositivo può essere utilizzato in sicurezza, richiedendo un protocollo formale, criteri di accettazione predefiniti e una documentazione completa.
- Verifica e validazione del software — verifica dei requisiti software e validazione del comportamento a livello di sistema secondo la norma IEC 62304. Per i dispositivi SaMD basati su intelligenza artificiale, la bozza di linee guida FDA di gennaio 2025 introduce requisiti specifici relativi alla valutazione del carico cognitivo, alla trasparenza e alla documentazione delle funzioni di override.
- Matrice di tracciabilità del progetto — collegamento di ogni requisito utente a una specifica ingegneristica verificata, garantendo che nessun requisito rimanga non testato.
- Rapporto di verifica del progetto (Design Verification Report, DVR) — raccolta strutturata di tutti i risultati delle attività di V&V, che costituisce il principale pacchetto di evidenze tecniche per le sottomissioni regolatorie.
La verifica e validazione non è una fase da comprimere sotto pressione temporale. Essa genera le evidenze su cui si basano le autorità regolatorie — e le lacune in questa fase rappresentano una delle principali cause di richieste di integrazione (deficiency letters) e ritardi nelle approvazioni.
Fase 4: Test preclinici
Prima che un dispositivo venga testato sull’uomo, deve superare una rigorosa valutazione preclinica per caratterizzarne la sicurezza biologica e funzionale. I test preclinici rispondono a una domanda fondamentale: questo dispositivo è sufficientemente sicuro da essere introdotto nel corpo umano?
In questa fase, prototipi sviluppati appositamente — non ancora dispositivi destinati alla produzione — vengono testati in ambienti di laboratorio controllati utilizzando metodi in vitro e in vivo. Gli studi su modelli animali rimangono una componente standard, ove appropriato in base al tipo di dispositivo, fornendo dati fisiologici che i test di laboratorio non possono replicare.
Attività principali:
- Test di biocompatibilità — le valutazioni secondo la serie ISO 10993 analizzano citotossicità, sensibilizzazione, genotossicità, tossicità sistemica, risposta all’impianto ed emocompatibilità. I test richiesti dipendono dal materiale del dispositivo, dal tipo di contatto (superficiale, comunicante con l’esterno o impiantabile) e dalla durata del contatto.
- Validazione della sterilizzazione — conferma dei livelli di garanzia di sterilità secondo ISO 11135 (ossido di etilene), ISO 11137 (radiazioni) o ISO 11607 (confezionamento), in funzione del metodo di sterilizzazione e del tipo di dispositivo.
- Sicurezza elettrica e compatibilità elettromagnetica (EMC) — secondo la norma IEC 60601-1 per dispositivi alimentati elettricamente, per garantire che il dispositivo non presenti rischi elettrici e non generi né subisca interferenze elettromagnetiche in ambienti ospedalieri.
- Validazione della durata di conservazione (shelf-life) e del confezionamento — dimostrazione che il dispositivo mantenga integrità strutturale, sterilità e prestazioni durante l’intera durata dichiarata, in condizioni definite di stoccaggio e trasporto.
- Tossicologia genetica e riproduttiva — valutazione del potenziale effetto cancerogeno o di eventuali impatti sulla salute riproduttiva, richiesta in particolare per dispositivi impiantabili o a contatto prolungato.
- Studi su modelli animali — quando necessario, gli studi in vivo forniscono dati su prestazioni del dispositivo, risposta tissutale e sicurezza, utili per l’ottimizzazione del design e la pianificazione degli studi clinici.
I dati preclinici confluiscono direttamente nelle sottomissioni regolatorie e costituiscono la base scientifica per la progettazione delle indagini cliniche. Carenze nei test preclinici possono ritardare o bloccare le richieste di autorizzazione IDE e i percorsi di marcatura CE.
Fase 5: Sperimentazione clinica
I dati preclinici, per quanto completi, non possono sostituire le evidenze cliniche sull’uomo. Le sperimentazioni cliniche generano dati di sicurezza e prestazione su pazienti reali e in contesti clinici reali e, per i dispositivi ad alto rischio, tali evidenze sono indispensabili per l’approvazione regolatoria.
Le sperimentazioni cliniche sui dispositivi medici sono indagini regolamentate dalla FDA ai sensi del 21 CFR 812, finalizzate alla raccolta di dati su sicurezza ed efficacia a supporto delle decisioni dell’autorità. Gli studi sono classificati come a rischio significativo, a rischio non significativo o esenti, con requisiti di IDE e approvazione dei comitati etici (IRB) proporzionati al livello di rischio.
L’Investigational Device Exemption (IDE) consente l’utilizzo di un dispositivo sperimentale in uno studio clinico per raccogliere dati su sicurezza ed efficacia. Le sperimentazioni cliniche sono generalmente condotte a supporto di una richiesta di approvazione PMA. Solo una piccola percentuale delle procedure 510(k) richiede dati clinici a supporto della domanda.
Attività principali:
- Piano di valutazione clinica (Clinical Evaluation Plan, CEP) — definisce la strategia per la generazione e l’analisi dei dati clinici in conformità al MDR (UE) 2017/745 o alle linee guida FDA. Il CEP stabilisce quali evidenze sono necessarie e come verranno raccolte, tramite studi prospettici, revisione della letteratura o utilizzo di dati di dispositivi equivalenti.
- Domanda IDE (USA) — tutte le valutazioni cliniche di dispositivi sperimentali, salvo esenzioni, richiedono un’IDE approvata prima dell’avvio dello studio. Per i dispositivi a rischio significativo, è necessaria anche l’approvazione della FDA.
- Progettazione dello studio clinico — definizione degli endpoint, della dimensione del campione, dei gruppi di controllo, dei periodi di follow-up e dei piani di analisi statistica. Il consorzio CORE-MD raccomanda un maggiore utilizzo di studi randomizzati controllati per dispositivi ad alto rischio, con comparatori attivi rappresentativi della migliore terapia disponibile.
- Approvazione del comitato etico (IRB/Comitato etico) — ottenimento dell’approvazione etica e definizione delle procedure di consenso informato prima dell’arruolamento dei soggetti.
- Conformità alle Good Clinical Practice (GCP) — le GCP rappresentano l’insieme di regolamenti e requisiti da rispettare nella conduzione degli studi clinici e si applicano a fabbricanti, sponsor, sperimentatori, comitati etici e al dispositivo stesso.
- Raccolta e analisi dei dati clinici — documentazione sistematica delle prestazioni del dispositivo, degli eventi avversi, degli esiti per gli utenti e di eventuali deviazioni dal piano di studio clinico.
- Rapporto di valutazione clinica (Clinical Evaluation Report, CER) — sintesi di tutte le evidenze cliniche — provenienti da studi, letteratura e dati di dispositivi equivalenti — per dimostrare che il dispositivo soddisfa i requisiti di sicurezza e prestazione ai fini dell’approvazione regolatoria.
La quantità e la profondità delle evidenze cliniche richieste aumentano proporzionalmente al rischio del dispositivo. Un dispositivo impiantabile di Classe III, primo della sua categoria, richiede anni di dati clinici prospettici. Un dispositivo di Classe II, a rischio inferiore, può soddisfare i requisiti attraverso una valutazione clinica basata sulla letteratura e una validazione limitata dei fattori umani. È fondamentale definire il percorso regolatorio prima di progettare la strategia di evidenza clinica.
Fase 6: Sottomissione e approvazione regolatoria
La sottomissione regolatoria è la fase in cui tutto il lavoro di sviluppo precedente — inclusi evidenze di progettazione, dati preclinici, evidenze cliniche e documentazione del sistema qualità — viene raccolto in una domanda formale per l’autorizzazione alla commercializzazione. Il percorso di sottomissione è determinato dalla classificazione del dispositivo e dal mercato di destinazione.
La FDA autorizza i dispositivi medici attraverso tre principali percorsi:
- 510(k) per dimostrare la sostanziale equivalenza
- De Novo per dispositivi innovativi a basso o medio rischio
- PMA (Premarket Approval) per la maggior parte dei dispositivi di Classe III
Nell’ambito del programma MDUFA V, gli obiettivi della FDA sono:
- 510(k): 95% delle decisioni entro 90 giorni FDA
- De Novo: 70% entro 150 giorni FDA
- PMA: tempo medio complessivo di decisione di circa 285 giorni nel periodo fiscale 2025–2027
Percorsi regolatori più comuni:
Mercato | Percorso | Classe del dispositivo |
USA (FDA) | 510(k) Clearance | Classe II |
USA (FDA) | De Novo Classification | Classe I/II (innovativi) |
USA (FDA) | PMA (Premarket Approval) | Classe III |
UE | Marcatura CE tramite fascicolo tecnico / design dossier | Classe I–III (MDR) |
Globale | MDSAP, TGA, Health Canada, PMDA | Specifico per mercato |
Documenti principali di sottomissione:
- Fascicolo tecnico / Design Dossier — documentazione completa di progettazione, test e produzione che dimostra la conformità ai requisiti applicabili.
- Fascicolo di gestione del rischio — documentazione conforme alla ISO 14971:2019 che copre l’intero processo di gestione del rischio, dall’identificazione dei pericoli fino all’accettabilità del rischio residuo.
- Rapporto di valutazione clinica (CER) — sintesi di tutte le evidenze cliniche a supporto delle dichiarazioni di sicurezza e prestazione del dispositivo.
- Evidenze del sistema qualità — certificazione ISO 13485:2016 o documentazione di conformità al QMSR secondo il 21 CFR Part 820 aggiornato, in vigore dal 2 febbraio 2026.
- Etichettatura e istruzioni per l’uso (IFU) — documentazione destinata all’utilizzatore finale, redatta in conformità ai requisiti normativi applicabili.
Un coinvolgimento proattivo con la FDA migliora significativamente i tempi e le probabilità di approvazione. La FDA fornisce feedback scritto entro 70 giorni di calendario dalla richiesta di un incontro pre-sottomissione. I team che interagiscono precocemente con le autorità regolatorie — tramite Q-Submission, incontri pre-IDE o programmi come il Breakthrough Device Designation, ove applicabili — gestiscono il processo di sottomissione in modo più efficiente e con un minor numero di criticità inattese.
Fase 7: Industrializzazione e validazione dei processi produttivi
L’approvazione regolatoria rappresenta una tappa fondamentale, non il traguardo finale. Prima che una singola unità commerciale venga distribuita, i processi produttivi devono dimostrare di essere in grado di produrre in modo costante dispositivi conformi alle specifiche, anche su scala industriale.
È necessario disporre di un sistema di gestione della qualità (QMS) per garantire il rispetto delle buone pratiche di fabbricazione. Ciò implica l’esecuzione di test e controlli sul dispositivo in diverse fasi del processo produttivo, prima della sua immissione sul mercato. Il controllo qualità è un’attività continua e il QMS deve essere conforme alla norma ISO 13485.
Attività principali:
- Installation Qualification (IQ) — verifica che le apparecchiature di produzione siano installate correttamente, calibrate e funzionino entro i parametri specificati prima dell’utilizzo.
- Operational Qualification (OQ) — conferma che i processi funzionino come previsto lungo l’intero intervallo operativo, comprese le condizioni limite.
- Performance Qualification (PQ) — dimostrazione della capacità di produrre in modo costante output conformi in condizioni reali di produzione, utilizzando personale, materiali e apparecchiature approvate su scala commerciale.
- Process FMEA (Failure Mode and Effects Analysis) — identificazione sistematica delle modalità di guasto nei processi produttivi e dei loro effetti sulla qualità del dispositivo, con implementazione di controlli per mitigare i rischi non accettabili prima dell’avvio della produzione.
- Qualifica dei fornitori — audit e approvazione formale dei fornitori di componenti critici e materie prime. Nel contesto del QMSR, i report di audit dei fornitori possono essere soggetti a ispezione da parte della FDA. La qualità dei fornitori è un’estensione diretta della qualità del dispositivo.
- Validazione delle camere bianche e del controllo della contaminazione — per dispositivi sterili o impiantabili, dimostrazione che gli ambienti controllati mantengano le classificazioni ambientali richieste dal processo produttivo.
La validazione dei processi è un requisito normativo previsto dal QMSR (21 CFR Part 820, che incorpora la ISO 13485:2016, clausola 7.5.6) e deve essere completata e documentata prima dell’inizio della distribuzione commerciale. Omettere o abbreviare questa fase comporta rischi di non conformità e aumenta la probabilità di guasti sul campo, deviazioni e richiami di prodotto.
Fase 8: Lancio commerciale
Il lancio commerciale rappresenta una delle sfide di coordinamento più complesse dell’intero programma di sviluppo. Produzione, supply chain, registrazioni regolatorie, infrastruttura commerciale, formazione clinica e sistemi post-commercializzazione devono essere tutti pienamente operativi contemporaneamente prima che il primo dispositivo raggiunga un paziente.
Attività principali:
- Registrazione del prodotto e autorizzazioni all’importazione — ottenimento delle autorizzazioni specifiche per ciascun Paese oltre all’approvazione principale. Molti mercati (ad esempio Cina – NMPA, Giappone – PMDA, Brasile – ANVISA) richiedono sottomissioni separate con documentazione tecnica locale.
- Configurazione dei canali di vendita e distribuzione — definizione della logistica, gestione delle scorte, evasione degli ordini e, ove necessario, gestione della catena del freddo per supportare la distribuzione commerciale.
- Formazione degli operatori sanitari (HCP) — sviluppo di programmi di formazione clinica, corsi pratici (in-service) e valutazioni delle competenze per gli utilizzatori. I materiali formativi devono essere coerenti con l’etichettatura approvata e le istruzioni per l’uso (IFU).
- Implementazione della strategia di rimborso — interazione con gli enti pagatori, definizione dei codici (CPT/HCPCS), conferma delle politiche di copertura e strutturazione dei meccanismi di rimborso. In assenza di rimborso, l’adozione sul mercato si blocca, indipendentemente dalle prestazioni cliniche del dispositivo.
- Attivazione del sistema di gestione dei reclami — implementazione di un sistema conforme a QMSR e MDR per la gestione degli eventi avversi prima della prima distribuzione. Gli obblighi post-commercializzazione iniziano dal momento in cui il dispositivo viene immesso sul mercato.
- Rilascio controllato sul mercato — avvio con una distribuzione geografica limitata per identificare eventuali problemi di produzione, prestazioni sul campo o formazione su scala gestibile prima dell’espansione commerciale completa. Un lancio controllato tutela sia i pazienti sia la reputazione del fabbricante.
Un lancio affrettato, guidato da tempistiche finanziarie piuttosto che dalla reale prontezza operativa, rappresenta una delle principali cause di problemi di qualità nelle fasi iniziali sul campo. Il lancio commerciale richiede lo stesso livello di rigore applicato in tutte le fasi precedenti.
Fase 9: Sorveglianza post-commercializzazione (PMS)
La sorveglianza post-commercializzazione non rappresenta la fine del processo di sviluppo, ma l’inizio di un obbligo continuo, lungo l’intero ciclo di vita del dispositivo, volto a monitorarne sicurezza e prestazioni nel mondo reale.
Il sistema PMS deve raccogliere, registrare e analizzare in modo attivo e sistematico dati rilevanti sulla qualità, sulle prestazioni e sulla sicurezza del dispositivo per tutta la sua durata. Tali dati devono essere utilizzati per determinare eventuali azioni correttive o preventive necessarie, nonché per monitorarne l’efficacia.
Per molte aziende di dispositivi medici operanti negli Stati Uniti e in Europa, la comprensione e la conformità a tre sistemi normativi — il MDR (UE) 2017/745, il QMSR della FDA (21 CFR Part 820, in vigore dal 2 febbraio 2026) e il nuovo quadro PMS del Regno Unito (in vigore dal 16 giugno 2025) — sono diventate fondamentali per garantire sia il successo commerciale sia la sicurezza dei pazienti.
Gli obblighi PMS previsti dal MDR europeo sono tra i più strutturati a livello globale. Il MDR richiede un sistema PMS formale e proattivo ai sensi degli articoli 83–86, integrato con il follow-up clinico post-commercializzazione (PMCF), la gestione del rischio, la valutazione clinica e il sistema di gestione della qualità ai sensi dell’articolo 10.
Frequenza dei report PSUR secondo MDR:
Classe del dispositivo | Requisito di reportistica | Frequenza |
Classe III e IIb | Periodic Safety Update Report (PSUR) | Almeno annuale |
Classe IIa | Periodic Safety Update Report (PSUR) | Almeno ogni 2 anni |
Classe I | Rapporto PMS | Secondo necessità / su richiesta |
L’ambito del PMCF è proporzionato al rischio. Un dispositivo impiantabile di Classe III, innovativo e ad alto rischio, può richiedere uno studio PMCF per tutta la durata del dispositivo, esteso su diversi anni. Un dispositivo di Classe I può invece richiedere un semplice questionario per raccogliere il feedback degli utenti, che includa comunque dati clinici relativi all’uso previsto e alle indicazioni.
Attività principali:
- Gestione dei reclami e analisi dei trend — monitoraggio di segnalazioni dal campo, registri di assistenza e resi per individuare segnali di sicurezza, pattern ricorrenti di guasto e degrado delle prestazioni. I dati PMS vengono utilizzati per identificare trend e, se necessario, segnalarli a EUDAMED. I requisiti di segnalazione dei trend sono disciplinati anche dalla vigilanza MDR (articolo 87).
- Segnalazione di incidenti (Medical Device Reporting / Vigilanza) — notifica degli eventi avversi gravi alla FDA, alle autorità competenti o agli organismi notificati entro i tempi previsti. Nell’UE, le azioni correttive di sicurezza sul campo (FSCA) devono essere comunicate agli utilizzatori tramite un avviso di sicurezza (Field Safety Notice, FSN).
- Follow-up clinico post-commercializzazione (PMCF) — processo continuo che aggiorna la valutazione clinica lungo l’intero ciclo di vita del dispositivo. Il rapporto PMCF documenta i risultati e alimenta sia il Clinical Evaluation Report (CER) sia il PSUR.
- Aggiornamento del fascicolo di gestione del rischio — integrazione dei dati di prestazione nel mondo reale, dei trend dei reclami e dei risultati PMCF nel fascicolo di gestione del rischio conforme alla ISO 14971. I dati PMS rappresentano il principale meccanismo per identificare nuovi pericoli dopo l’immissione sul mercato.
- Summary of Safety and Clinical Performance (SSCP) — documento pubblico richiesto per dispositivi di Classe III e impiantabili nell’UE, aggiornato in linea con il PSUR e pubblicato in EUDAMED.
- Azioni correttive sul campo e richiami — esecuzione di correzioni, aggiornamenti software o ritiri dal mercato quando vengono identificati problemi di sicurezza. Azioni tempestive e documentazione completa sono obblighi sia normativi sia di sicurezza per i pazienti.
I dati PMS chiudono il ciclo tra prestazioni sul mercato e sviluppo del prodotto. I trend dei reclami guidano il design delle generazioni successive. I risultati PMCF aggiornano la valutazione clinica. Il fascicolo di gestione del rischio evolve con l’accumularsi delle evidenze reali. I fabbricanti che considerano il PMS come un sistema attivo di intelligence, e non come un mero adempimento normativo, sviluppano dispositivi più sicuri, rispondono più rapidamente ai problemi e costruiscono relazioni più solide con le autorità regolatorie nel tempo.
Il processo di sviluppo dei dispositivi medici è lungo, complesso e non tollera scorciatoie. Tuttavia, il rigore che richiede ha un unico obiettivo: garantire che le persone che dipendono da questi dispositivi possano contare su una sicurezza ed efficacia dimostrate da evidenze solide.