
Medical device manufacturing is one of the most tightly regulated and technically demanding industries in the world. From the surgical tools used in operating rooms to the pacemakers keeping hearts beating, every device must be designed, produced, and tested to exacting standards before it ever reaches a patient.
This guide covers everything you need to know about how medical devices are made — starting with what the industry actually involves and how devices are classified by risk level into Class I (low risk), Class II (moderate risk), and Class III (high risk) categories. Each class carries different regulatory obligations and manufacturing requirements.
We walk through the full design and development process, from identifying a clinical need and building early prototypes to risk management, design verification, and clinical validation. We then cover the core manufacturing workflow — material selection, component fabrication, assembly, quality control, sterilization, packaging, and final distribution — and explain the key technologies that make it possible, including CNC machining, injection molding, 3D printing, robotics, cleanroom environments, and advanced inspection systems.

Quality control is examined in depth, covering incoming material inspection, in-process monitoring, functional testing, biocompatibility assessment, sterility verification, and post-market surveillance. On the regulatory side, we break down the global frameworks manufacturers must navigate — including FDA requirements, EU MDR, and ISO 13485 — along with the key documents every manufacturer must maintain: the Design History File, Device Master Record, and Device History Record.
We also explore the five biggest challenges facing the industry today, the trends shaping its future — from AI and digital twins to sustainable and decentralized manufacturing — and answer the most frequently asked questions about timelines, approvals, and compliance.
What is Medical Device Manufacturing?

Medical device manufacturing is the process of designing, producing, assembling, testing, and distributing devices used in healthcare settings. Because these products are used directly in patient care — from surgical tools and pacemakers to diagnostic imaging equipment and wearable health technologies — the process is highly precise and strictly regulated to ensure patient safety and product reliability.
The industry produces a wide range of products, including basic medical instruments, advanced technological equipment, implantable devices, patient monitoring systems, and wearable health technologies. Because they are used directly in patient care, manufacturers must follow strict quality standards and regulatory guidelines throughout the entire production lifecycle.
The process begins with research and product design, where engineers, designers, and healthcare professionals collaborate to address specific clinical needs. After the design phase, manufacturers select appropriate raw materials and produce individual components using specialized techniques. These are then assembled into finished devices within controlled production environments. Every device must pass rigorous quality control — including functional testing, sterility verification, biocompatibility assessments, and durability evaluations — before it can be approved for clinical use.
Overall, medical device manufacturing combines engineering expertise, advanced production technologies, quality management systems, and regulatory compliance to produce devices that support modern healthcare and improve patient outcomes.
What Are the Types of Medical Devices?
Medical devices come in many forms and are generally categorized based on their purpose, function, and level of risk to patients. Regulatory authorities around the world classify these devices to ensure that the appropriate safety standards, testing procedures, and approval processes are applied before they are used in healthcare environments.
Most regulatory frameworks group medical devices into three main risk-based categories: Class I, Class II, and Class III. Each class represents a different level of potential risk to patients and therefore requires different levels of regulatory oversight.
Class I Devices (Low Risk)
Class I medical devices are considered low-risk products and usually have simple designs with minimal potential for harm to patients. These devices typically do not require extensive regulatory approval and are subject to general quality and safety controls.
Examples of Class I devices include:
- Bandages
- Surgical gloves
- Manual stethoscopes
- Tongue depressors
- Hospital beds
Although these devices are relatively simple, manufacturers must still ensure they are produced according to quality management standards and proper manufacturing practices.
Class II Devices (Moderate Risk)
Class II devices present a moderate level of risk and usually require additional regulatory controls to ensure their safety and effectiveness. These devices often involve more complex designs or technologies and may require clinical data, performance testing, and regulatory review before they can be marketed.
Examples of Class II devices include:
- Infusion pumps
- Blood pressure monitors
- Diagnostic imaging equipment
- Powered wheelchairs
- Pregnancy test kits
Manufacturers of Class II devices must follow stricter manufacturing processes and quality assurance procedures to meet regulatory requirements.
Class III Devices (High Risk)
Class III medical devices are considered high-risk devices because they are typically used to support or sustain life, are implanted in the body, or pose significant risk if they fail. These devices require the highest level of regulatory scrutiny, including extensive clinical testing and approval before they can be sold.
Examples of Class III devices include:
- Pacemakers
- Artificial heart valves
- Implantable defibrillators
- Deep brain stimulators
Due to their critical role in patient health, Class III devices must undergo rigorous design validation, safety testing, and regulatory review before entering the market.
Other Ways to Categorize Medical Devices
Beyond risk-based classification, medical devices can also be grouped by their function or intended use. Some common categories include:
- Diagnostic devices – used to detect or monitor diseases
- Therapeutic devices – used to treat medical conditions
- Surgical instruments – tools used during medical procedures
- Monitoring devices – equipment that tracks patient health data
Understanding these categories helps manufacturers, healthcare providers, and regulators determine the appropriate manufacturing processes, safety requirements, and regulatory pathways needed to bring safe and effective medical devices to market.
How Are Medical Devices Designed and Developed?

The design and development of medical devices is a structured, multi-stage process that transforms a clinical need into a safe, functional, and regulatory-compliant product. Because medical devices directly affect patient health, the development process must follow strict engineering practices, risk management principles, and regulatory design controls.
Typically, medical device development begins with identifying a clinical problem or unmet healthcare need. Engineers, clinicians, and product designers collaborate to define the device’s intended use, performance requirements, and user environment. This stage often involves market research, clinical input, and feasibility studies to determine whether the concept is technically and commercially viable.
Concept Development and Design Planning
Once the need is clearly defined, the design team begins concept development and planning. During this stage, engineers outline the device’s specifications, materials, technical architecture, and performance criteria. Design planning also includes creating a development timeline, assigning responsibilities, and defining regulatory requirements that must be met throughout the project.
This stage ensures that the product is designed according to medical device design control standards, which are required by regulatory bodies to maintain traceability and quality throughout development.
Prototyping and Engineering Design
After the initial concept is approved, the next step is prototyping and engineering design. Engineers create early versions of the device using techniques such as computer-aided design (CAD), simulation tools, and rapid prototyping technologies like 3D printing.
Prototypes allow teams to evaluate important aspects of the device, including:
- functionality
- ergonomics and usability
- mechanical performance
- electrical integration (for electronic devices)
Multiple design iterations are typically developed and tested before a final design is selected.
Risk Management and Design Verification
Medical device development also requires extensive risk analysis and design verification. Manufacturers must identify potential hazards associated with the device and implement controls to minimize risks to patients and healthcare providers.
Verification activities confirm that the device design meets the specified engineering requirements. These activities may include mechanical testing, electrical safety testing, software validation, and performance evaluations.
Risk management processes often follow internationally recognized frameworks such as ISO 14971, which focuses on identifying, analyzing, and mitigating potential device risks.
Design Validation and Clinical Evaluation
After verification confirms the design meets technical specifications, manufacturers perform design validation to ensure the device performs safely and effectively in real-world conditions.
Validation activities may involve:
- usability testing with healthcare professionals
- simulated clinical environments
- human factors engineering studies
- clinical trials (for higher-risk devices)
These evaluations confirm that the device meets the needs of its intended users and functions properly in clinical settings.
Preparing for Manufacturing
Once the device design is validated, manufacturers finalize the design documentation and production specifications needed for large-scale manufacturing. This includes defining materials, production processes, assembly instructions, quality control procedures, and packaging requirements.
At this stage, the device moves from the development phase into regulated manufacturing and regulatory submission, where it undergoes formal approval processes before entering the market.
The Importance of Design Controls
Throughout the entire design and development process, manufacturers must maintain detailed documentation known as design controls. These controls track every stage of development—from initial concept to final design—ensuring traceability, quality assurance, and regulatory compliance.
By following a structured design and development process, medical device manufacturers can create products that are safe, reliable, effective, and compliant with global healthcare regulations, ultimately improving patient care and medical outcomes.
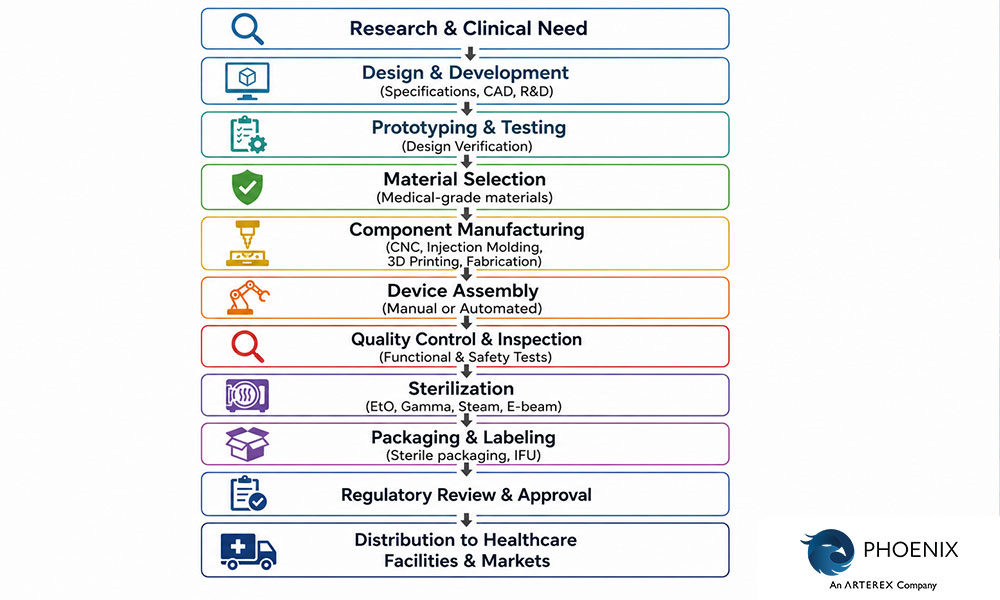
What is the Medical Device Manufacturing Process?
The medical device manufacturing process refers to the series of controlled steps used to produce medical devices from raw materials to finished products ready for clinical use. Because these devices directly affect patient health and safety, the manufacturing process must follow strict quality standards, validated production methods, and regulatory requirements.
While the exact workflow varies depending on the type of device being produced, most medical devices go through several core manufacturing stages, including material preparation, component fabrication, assembly, testing, sterilization, and packaging. Each step must be carefully monitored to ensure the final product meets safety, performance, and regulatory expectations.
1. Material Selection and Preparation
The process begins with selecting appropriate raw materials that meet medical-grade standards. These materials must be safe, durable, and compatible with the device’s intended use. Common materials used in medical device manufacturing include medical-grade polymers, stainless steel, titanium alloys, ceramics, and specialized electronic components.
Manufacturers must also verify that materials meet biocompatibility and regulatory requirements, especially for devices that come into direct contact with the human body.
2. Component Manufacturing
Once materials are selected, individual parts of the device are produced through various fabrication techniques. The specific manufacturing method depends on the design and function of the device.
Common techniques include:
- CNC machining for precision metal components
- Injection molding for plastic parts
- Laser cutting and microfabrication for small or intricate components
- Additive manufacturing (3D printing) for complex geometries
These processes allow manufacturers to create highly precise components that meet strict engineering specifications.
3. Device Assembly
After individual components are produced, they are assembled into the final medical device. Assembly may involve manual operations performed by trained technicians or automated processes using robotic systems.
For devices that include electronics or sensors, assembly may also involve circuit integration, software installation, and calibration to ensure the device functions correctly.
4. Quality Control and Inspection
Quality assurance is a critical part of medical device manufacturing. Throughout the production process, manufacturers perform inspections and tests to verify that each component and assembled device meets design specifications and safety standards.
Common quality control activities include:
- visual inspections
- dimensional measurements
- functional testing
- electrical safety testing
- performance verification
These checks help identify and correct defects before the device reaches healthcare providers.
5. Sterilization
Many medical devices must be sterilized to eliminate microorganisms before they can be used in clinical settings. The sterilization method depends on the materials and design of the device.
Common sterilization methods include:
- steam sterilization (autoclaving)
- ethylene oxide (EtO) sterilization
- gamma radiation sterilization
- electron beam sterilization
Sterilization processes must be validated to ensure they effectively eliminate pathogens without damaging the device.
6. Packaging and Labeling
Once sterilized, devices are packaged in protective, sterile packaging that maintains product integrity during storage and transportation. Packaging is designed to prevent contamination, physical damage, and environmental exposure.
Manufacturers must also include proper labeling that provides important information such as instructions for use, safety warnings, manufacturing details, and regulatory compliance markings.
7. Final Testing and Distribution
Before devices are shipped, they undergo final verification and documentation review to confirm that all manufacturing and quality requirements have been met. After approval, the products are distributed to hospitals, clinics, and healthcare providers.
Throughout the entire manufacturing process, companies must maintain detailed records and follow quality management systems to ensure traceability, consistency, and compliance with global medical device regulations.
In summary, the medical device manufacturing process is a highly controlled and systematic workflow designed to ensure that every device produced is safe, reliable, and effective for patient care.
Medical Device Manufacturing Process Flow Chart
What Technologies Are Used in Medical Device Manufacturing?
Medical device manufacturing relies on a wide range of advanced technologies and precision engineering methods to produce safe, reliable, and high-performance healthcare products. Because medical devices often involve complex designs and strict regulatory requirements, manufacturers must use specialized production technologies that ensure accuracy, consistency, and quality throughout the manufacturing process.
From automated machinery to digital design tools, modern medical device production integrates mechanical engineering, materials science, electronics, and software technologies to create devices that meet demanding clinical standards.
Computer-Aided Design (CAD) and Simulation
One of the most important technologies used during the development stage is computer-aided design (CAD). Engineers use CAD software to create detailed digital models of medical devices, allowing them to visualize the product, test design variations, and identify potential design issues before physical prototypes are built.
Simulation tools are often used alongside CAD to evaluate factors such as mechanical strength, heat resistance, fluid dynamics, and electrical performance. This helps manufacturers optimize device designs while reducing development time and production costs.
CNC Machining
Computer Numerical Control (CNC) machining is widely used to manufacture precise components for medical devices. CNC machines operate through computer-controlled instructions that guide cutting tools to shape metal or plastic materials into exact dimensions.
This technology is commonly used to produce components such as surgical instruments, orthopedic implants, and device housings. CNC machining provides high accuracy and repeatability, which are essential for medical products that must meet strict tolerances.
Injection Molding
Injection molding is a common manufacturing technique used to produce plastic components in large quantities. In this process, molten plastic is injected into a mold cavity where it cools and solidifies into the desired shape.
Many disposable medical products—such as syringes, IV connectors, and diagnostic cartridges—are produced using injection molding. The process allows manufacturers to produce complex shapes with high consistency and efficiency.
Additive Manufacturing (3D Printing)
Additive manufacturing, commonly known as 3D printing, has become increasingly important in the medical device industry. This technology builds components layer by layer using digital models, allowing manufacturers to create highly complex geometries that may be difficult or impossible to produce with traditional methods.
3D printing is often used for:
- rapid prototyping
- customized implants and prosthetics
- surgical guides and models
- small-batch production of specialized devices
This technology is particularly valuable for creating patient-specific medical devices.
Robotics and Automation
Robotic systems and automated production lines are widely used in modern medical device manufacturing to improve efficiency and precision. Automation helps reduce human error and ensures consistent production quality.
Robotic systems can perform tasks such as:
- component assembly
- precision welding
- adhesive dispensing
- device inspection
Automation is especially important for manufacturing electronic medical devices and complex assemblies.
Laser Manufacturing and Microfabrication
Laser-based technologies are used for high-precision cutting, welding, and marking in medical device production. Lasers allow manufacturers to work with extremely small components without damaging delicate materials.
Laser processing is commonly used in the fabrication of:
- surgical instruments
- stents and vascular devices
- microelectronic components
- medical device labeling and traceability markings
Cleanroom Manufacturing
Many medical devices must be produced in controlled cleanroom environments to prevent contamination from dust, microbes, or airborne particles. Cleanrooms are specially designed facilities where air quality, temperature, humidity, and particle levels are carefully controlled.
Cleanroom manufacturing is essential for devices such as implants, diagnostic cartridges, and sterile medical products.
Advanced Inspection and Quality Technologies
To ensure product safety and compliance, manufacturers also use advanced inspection and testing technologies. These tools help detect defects, verify dimensions, and confirm device performance.
Examples include:
- automated optical inspection systems
- coordinate measuring machines (CMM)
- X-ray and imaging inspection
- electronic testing equipment
Emerging technologies including artificial intelligence, digital twins, and smart Industry 4.0 manufacturing systems are increasingly transforming how medical devices are designed, produced, and monitored, opening new possibilities for quality assurance and operational efficiency.
How is Quality Control and Testing Performed in Medical Device Manufacturing?

Quality assurance in medical device manufacturing is managed through a Quality Management System (QMS) that governs every stage of production. Key activities include:
- Incoming Material Inspection — Raw materials and purchased components are verified for composition, dimensional accuracy, supplier certifications, and regulatory compliance.
- In-Process Quality Control — Dimensional measurements, visual inspections, equipment calibration checks, and process validation monitoring are conducted during production to catch issues early.
- Functional & Performance Testing — Assembled devices undergo electrical safety testing, mechanical stress testing, software validation, and calibration checks under simulated real-world conditions.
- Biocompatibility Testing — Devices contacting the human body are tested per ISO 10993 for cytotoxicity, sensitization, irritation, genotoxicity, and other biological responses.
- Sterility & Contamination Testing — Microbial contamination testing, sterilization validation, and cleanroom environmental monitoring ensure devices are safe for clinical use.
- Final Inspection & Release — A comprehensive review of finished devices, packaging integrity, labeling, and manufacturing documentation confirms compliance before distribution.
- Post-Market Surveillance — Manufacturers monitor field performance, track complaints and adverse events, and implement Corrective and Preventive Actions (CAPA) to maintain ongoing patient safety.
What Are the Regulatory Requirements for Medical Device Manufacturing?
Medical device manufacturing is subject to strict regulatory oversight across the world to ensure that medical technologies are safe, effective, and consistently produced. Because medical devices directly affect patient health, governments and regulatory agencies require manufacturers to follow detailed rules governing device design, production processes, quality management, clinical evaluation, and post-market monitoring.
Although regulations vary by region, most regulatory systems share common principles. These include risk-based device classification, quality management systems, regulatory approval pathways, and continuous monitoring after the device reaches the market.
Global Regulatory Frameworks
Medical device manufacturers must comply with the regulations of the specific markets where their products are sold. Each region has its own regulatory authority responsible for reviewing devices, inspecting manufacturing facilities, and granting market authorization.
The table below summarizes major global regulatory bodies and their key regulatory frameworks.
Region | Regulatory Body | Primary Regulation | Market Access Document |
United States | FDA (Center for Devices and Radiological Health – CDRH) | 21 CFR Parts 800–898 | 510(k), PMA, De Novo |
European Union | Notified Bodies under EU MDR | EU MDR 2017/745 | CE Marking |
United Kingdom | Medicines and Healthcare products Regulatory Agency (MHRA) | UK MDR 2002 (amended) | UKCA Marking |
Canada | Health Canada | Canadian Medical Devices Regulations (CMDR) | Medical Device License |
Australia | Therapeutic Goods Administration (TGA) | Therapeutic Goods Act 1989 | ARTG Inclusion |
Japan | Pharmaceuticals and Medical Devices Agency (PMDA) | Pharmaceutical and Medical Device Act (PAL/MPAL) | Shonin / Ninsho Approval |
China | National Medical Products Administration (NMPA) | Regulations on Supervision of Medical Devices | NMPA Registration |
Brazil | National Health Surveillance Agency (ANVISA) | RDC 751/2022 | ANVISA Registration |
These regulatory frameworks define how medical devices must be evaluated, approved, and monitored before and after entering the market.
Quality Management System Requirements
Most regulatory authorities require manufacturers to implement a Quality Management System (QMS) to ensure that production processes are controlled and consistent. A QMS defines the procedures used to manage product design, supplier quality, production controls, documentation, and corrective actions.
Many regulatory systems align with ISO 13485, an international standard that specifies requirements for quality management in medical device manufacturing. This standard helps manufacturers maintain traceability, risk management practices, and continuous quality improvement.
Premarket Approval and Device Registration
Before a medical device can be sold, manufacturers must submit documentation to regulatory authorities demonstrating that the device is safe and performs as intended. The approval pathway typically depends on the risk classification of the device.
For example:
- In the United States, manufacturers may submit a 510(k) notification, De Novo classification request, or Premarket Approval (PMA) depending on device risk.
- In the European Union, manufacturers must obtain a CE Mark after undergoing conformity assessment under the EU MDR.
- In countries like Canada, Australia, and Japan, manufacturers must obtain official device licenses or approvals before marketing their products.
These processes require manufacturers to provide technical documentation such as device design information, risk assessments, testing results, and clinical evidence.
Manufacturing Controls and Documentation
Regulatory requirements also emphasize process validation and documentation. Manufacturers must maintain records covering all stages of production, including material sourcing, component manufacturing, assembly procedures, and testing results.
Key documentation may include:
- design history files
- device master records
- batch production records
- inspection and testing reports
- supplier qualification documentation
These records ensure full traceability of each medical device produced.
Post-Market Surveillance
Regulatory compliance does not end when the device is approved. Manufacturers must continuously monitor product performance after market release through post-market surveillance systems.
These systems involve:
- reporting adverse events and safety incidents
- monitoring product complaints
- conducting recalls or corrective actions if necessary
- collecting real-world performance data
Post-market monitoring helps regulators identify potential safety issues and ensures that medical devices remain safe throughout their lifecycle.
Regulatory requirements play a critical role in protecting patients and maintaining trust in healthcare technologies. By following global regulations and quality standards, medical device manufacturers can ensure their products are safe, reliable, and compliant with international healthcare regulations, allowing them to reach global markets while maintaining the highest standards of patient care.
What Are the Challenges in Medical Device Manufacturing?
Medical device manufacturing involves complex processes, strict regulations, and advanced technologies. While the industry continues to innovate rapidly, manufacturers must overcome several major challenges to ensure devices are safe, effective, and commercially viable. The following are the five most significant challenges faced by medical device manufacturers.
1. Regulatory Compliance and Changing Regulations
Medical device manufacturers must comply with multiple regulatory frameworks across global markets. Each region has its own approval pathways, documentation requirements, and inspection standards.
Regulations also evolve over time, such as updates to medical device laws or new safety guidelines. Keeping up with regulatory changes while maintaining compliance can be time-consuming and resource-intensive for manufacturers.
2. High Development and Production Costs
Developing a medical device often requires significant investment in research, engineering, prototyping, clinical testing, and regulatory approval. These costs can increase further when manufacturers must perform extensive validation, certification, and quality testing.
For startups and smaller companies, managing these costs while remaining competitive in the market can be a major challenge.
3. Maintaining Strict Quality and Safety Standards
Medical devices must meet extremely strict quality, safety, and performance requirements. Even small defects can pose risks to patients and lead to regulatory actions or product recalls.
Manufacturers must implement comprehensive quality management systems, rigorous testing procedures, and detailed documentation to ensure that every device meets required standards.
4. Complex Supply Chain Management
Medical device production often depends on specialized components, medical-grade materials, and highly qualified suppliers. Managing this supply chain can be difficult, particularly when suppliers operate across different countries.
Disruptions in raw materials, electronic components, or logistics can delay production schedules and increase manufacturing costs.
5. Rapid Technological Change
The healthcare industry is evolving quickly with innovations such as robotic surgery, wearable health devices, AI-powered diagnostics, and smart medical technologies. Manufacturers must continuously upgrade their manufacturing capabilities and technical expertise to keep pace with these advancements.
Adapting to new technologies while maintaining regulatory compliance and product quality can be a significant operational challenge.
What Are the Future Trends in Medical Device Manufacturing?
Medical device manufacturing is entering a period of transformation unlike anything the industry has experienced before. Converging advances in materials science, digital technology, artificial intelligence, and biotechnology are simultaneously redefining what devices can do, how they are made, and how they are regulated.
Here is a comprehensive look at the trends shaping the future of medical device manufacturing.
Trend | Core Opportunity | Primary Manufacturing Impact |
AI and Machine Learning | Faster development, better quality control | Inspection automation, predictive systems |
Personalized Devices | Superior clinical outcomes, premium markets | Patient-specific production models |
Advanced Additive Manufacturing | New geometries, on-demand production | Process validation, point-of-care manufacturing |
Digital Twins | Virtual testing, process optimization | Reduced validation burden, closed-loop control |
Industry 4.0 / Smart Manufacturing | Real-time visibility, paperless operations | MES integration, IIoT infrastructure |
Advanced Materials | New clinical capabilities, reduced interventions | New process qualification requirements |
Minimally Invasive / Robotic Devices | Clinical demand growth, premium positioning | Miniaturization, high-precision assembly |
Wearables and Connected Devices | Continuous monitoring, chronic disease management | Consumer-medical manufacturing convergence |
Sustainable Manufacturing | Regulatory compliance, brand differentiation | Material substitution, packaging redesign |
Decentralized Manufacturing | Supply resilience, market responsiveness | Distributed networks, on-demand production |
Regulatory Science Evolution | Faster pathways, computational evidence | Digital submission infrastructure |
Biologic-Device Convergence | Regenerative medicine, cell therapy | Aseptic biological manufacturing capability |
The future of medical device manufacturing will be defined not by any single technology, but by the ability to integrate many technologies simultaneously — connecting digital design to intelligent manufacturing, personalized production to scalable quality systems, and biological innovation to regulatory frameworks capable of evaluating it responsibly.
The manufacturers who will lead this future are those building those integrations today — not waiting for the technology to mature or the regulation to catch up, but investing in capability, culture, and infrastructure that positions them to move quickly when the moment arrives.
The patient waiting for the next generation of devices cannot afford for that moment to be delayed.
Frequently Asked Questions
How long does it take to bring a medical device to market?
Device Class | Typical Timeline |
Class I (low risk) | 6 months – 1 year |
Class II (moderate risk, 510(k)) | 1 – 3 years |
Class III (high risk, PMA) | 4 – 7 years (up to a decade with clinical trials) |
What regulations govern medical device manufacturing?
The key frameworks include FDA 21 CFR Part 820 / QMSR (U.S.), EU MDR 2017/745 (Europe), EU IVDR 2017/746 (in vitro diagnostics), ISO 13485 (global QMS standard), ISO 14971 (risk management), IEC 60601 (electrical medical equipment safety), and IEC 62304 (medical device software). Most manufacturers must comply with several of these simultaneously.
What is ISO 13485 and why does it matter?
ISO 13485 defines the Quality Management System requirements specific to medical device design, development, production, delivery, and post-market monitoring. Certification is a market access requirement in the EU, Canada, Australia, Japan, and many other countries. Without it, manufacturers cannot legally sell in most major international markets or win contracts with large OEMs and hospital systems.
What is a 510(k) clearance?
A 510(k) is the FDA pathway for Class II devices. It allows market entry by demonstrating substantial equivalence to an already-approved predicate device, rather than conducting full clinical trials. Manufacturers must still provide technical documentation, performance data, biocompatibility evidence, and labeling. FDA review typically takes 3–12 months.
What is Premarket Approval (PMA)?
PMA is the most rigorous FDA pathway, required for Class III devices. It demands valid scientific evidence — typically including clinical trial data — proving the device is both safe and effective. The process can take 2–5 years and cost tens of millions of dollars. Examples of PMA-approved devices include implantable cardiac defibrillators and deep brain stimulation systems.
What is Good Manufacturing Practice (GMP)?
GMP refers to the regulations and procedures ensuring medical devices are consistently produced to a quality standard appropriate for their intended use. In the U.S., it is defined by 21 CFR Part 820; internationally, ISO 13485 governs equivalent requirements. GMP covers facility design, equipment qualification, personnel training, documentation, process validation, nonconforming product management, CAPA systems, and complaint handling.
What is process validation?
Process validation is documented evidence that a manufacturing process consistently produces a product meeting its specifications when operated within defined parameters. The three stages are: Installation Qualification (IQ) — confirming equipment is installed correctly; Operational Qualification (OQ) — confirming equipment operates as intended; and Performance Qualification (PQ) — confirming the process consistently produces acceptable product under real production conditions.
What are the Design History File (DHF), Device Master Record (DMR), and Device History Record (DHR)?
- DHF — A compilation of records describing the complete design history of a device, required by FDA 21 CFR Part 820.30 for Class II and III devices. It includes design inputs, outputs, review records, verification and validation results, and change records.
- DMR — The complete set of specifications and instructions required to manufacture a device: device specs, production procedures, quality assurance procedures, and packaging/labeling specifications. Every unit must be produced in accordance with the DMR.
- DHR — The production record for each manufactured unit or batch, proving it was built to the DMR. It contains dates, quantities, lot numbers, acceptance results, labels used, and operator identities — making every device fully traceable.
What does ‘biocompatibility’ mean?
Biocompatibility refers to a material’s or device’s ability to perform its intended function without causing toxic, injurious, or immunological responses in patients. It is evaluated according to ISO 10993, with testing protocols determined by the nature, duration, and degree of patient contact. Tests include cytotoxicity, sensitization, irritation, systemic toxicity, genotoxicity, and implantation testing, all of which must be documented before regulatory approval.
- What is Medical Device Manufacturing?
- What Are the Types of Medical Devices?
- How Are Medical Devices Designed and Developed?
- What is the Medical Device Manufacturing Process?
- What Technologies Are Used in Medical Device Manufacturing?
- How is Quality Control and Testing Performed in Medical Device Manufacturing?
- What Are the Regulatory Requirements for Medical Device Manufacturing?
- What Are the Challenges in Medical Device Manufacturing?
- What Are the Future Trends in Medical Device Manufacturing?
- Frequently Asked Questions